To maximize Health Care Resources, advance the world’s leading Research and to provide the premium support for all the Australasian Leukodystrophy families with the outcome being a cure.
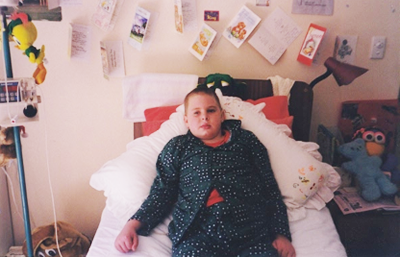
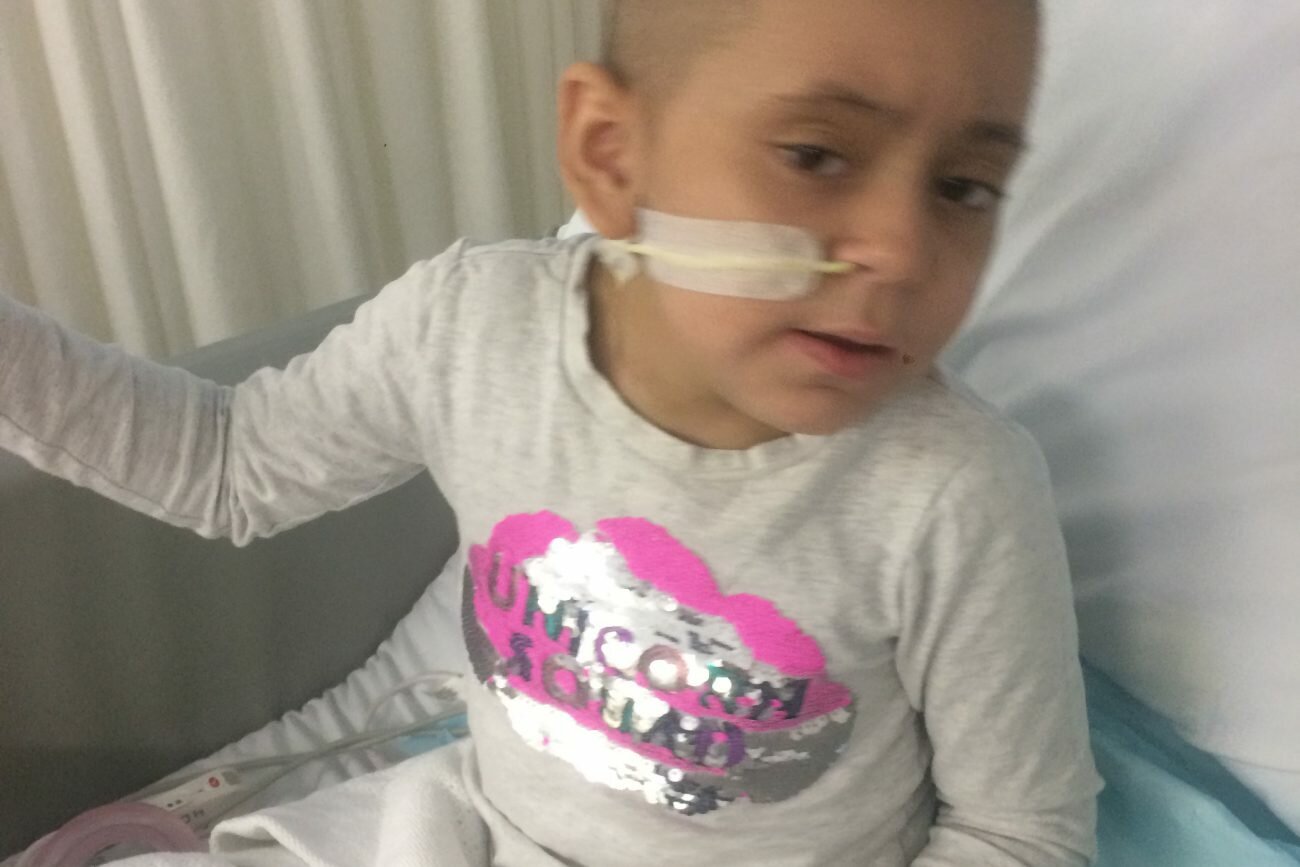
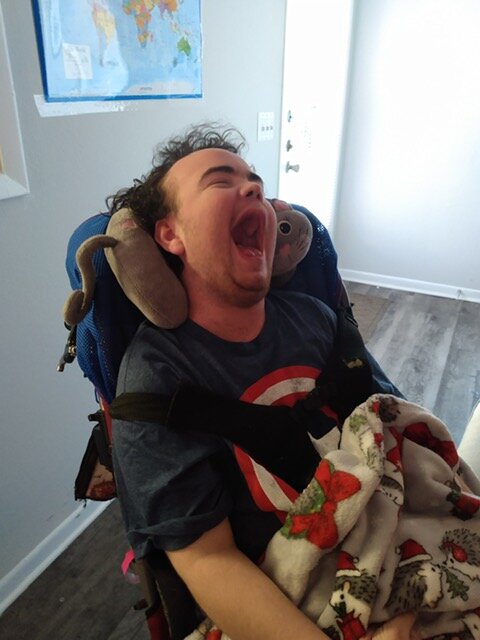
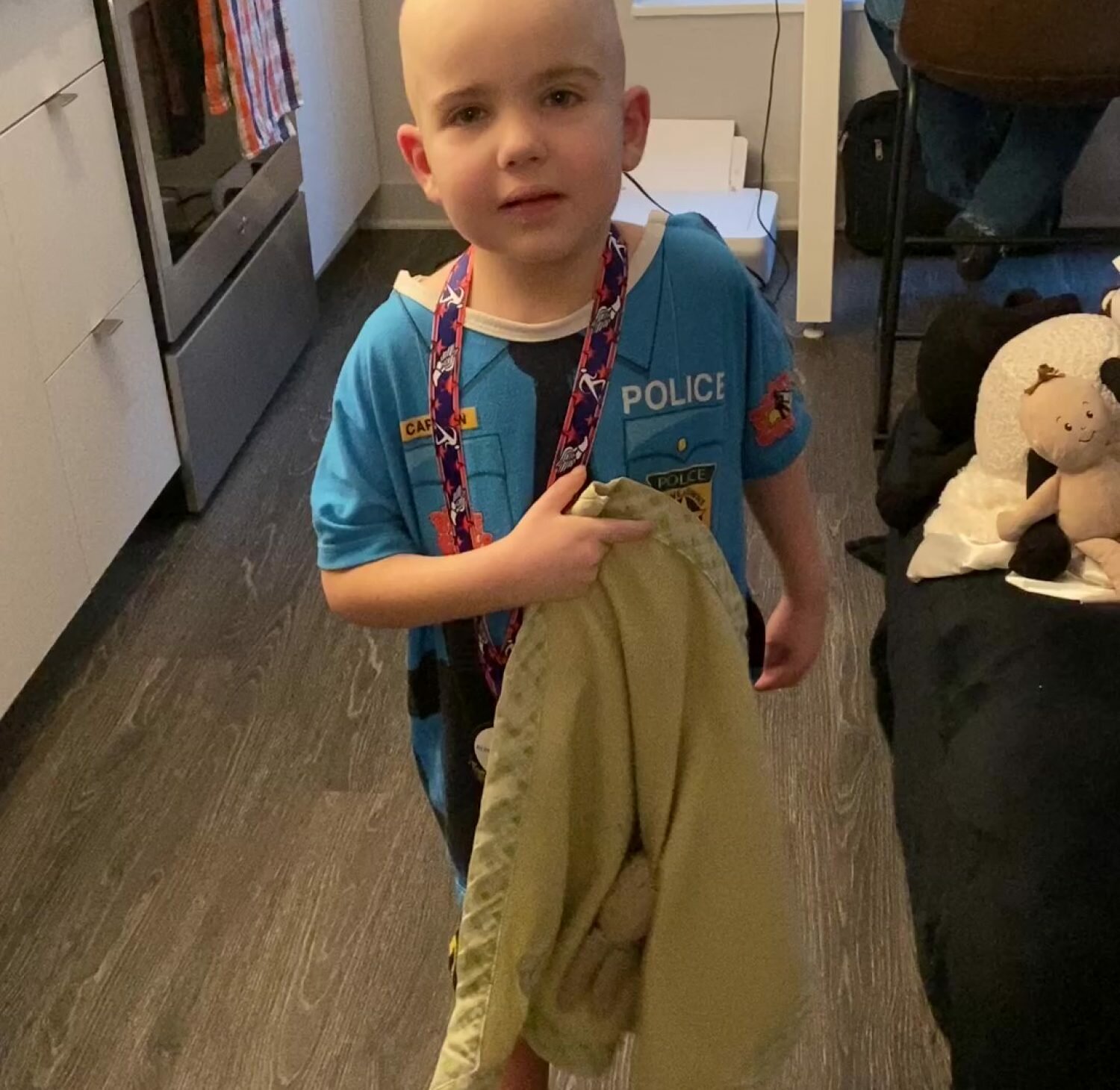
Brendan Wyborn was born in Townsville Queensland Australia on the 7th of March 1988. His life was as any normal little boy until he was about 8 years old. He moved to live in Hervey Bay Queensland and enjoyed fishing, playing cricket, his older Sister Emma and of course his family and friends.
The term Leukodystrophy refers to a group of genetic disorders that are characterized by the imperfect growth, development or maintenance of the white matter which is known as myelin.
Adrenoleukodystrophy added to the Recommended Uniform Screening Panel
We are very happy to announce that on February 17th, 2016, the US Secretary of the Department of Health u0026amp; Human Services, Sylvia Burwell, approved the addition of adrenoleukodystrophy (ALD) to the Recommended Uniform Screening Panel (RUSP), the federal list of genetic diseases recommended for state newborn screening programs. Read her letter here.
The addition of ALD to the RUSP will expedite the process of adding ALD to state newborn screening programs. California and New Jersey can now join New York and Connecticut in screening newborns for ALD allowing for timely diagnosis and treatment. Our work has only begun, however: there are still 46 states in the US that need to begin ALD newborn screening as soon as possible. In Australia we are undertaking a lot of work to achieve the same result here, hopefully later this year.
We would like to thank Secretary Burwell, the incredible team of ALD advocates across the world, and to all of you for your support to help make this vision a reality.